
In the digital era, where the volume of data is growing exponentially, understanding storage solutions becomes pivotal, especially in fields like bioinformatics that deal with enormous datasets. One term that often crops up when discussing modern storage solutions is “cloud buckets”. But what are they? And how do they propel the advances in bioinformatics?
When someone talks about “cloud buckets”, more often than not they are referring to a type of storage called object storage. Object storage is inherently different from traditional file system storage. Your traditional file system storage manipulates and manages files (data) in a hierarchical folder structure through your operating system (OS) with fixed system attributes and is generally accessed through a single site. Object storage is a storage system that manages and manipulates data storage as distinct units called objects in a flat namespace using an API rather than an OS. Each object has a unique ID, data, metadata, and attributes which are not fixed by an OS.
So what are cloud buckets? They are virtual containers for object storage. When referring to cloud buckets for bioinformatics, curators of data will often store related objects within the same buckets. This relatedness often involves permissions and specific types of data. For example, sequence data repositories will often provide bucket access to genomic data. They could provide public access (permissions) to assembled genomes (related data) through a Google or AWS bucket.
Note: It’s important to remember that cloud buckets are NOT file systems. They often will appear to have file paths, but these are virtual file paths by adding a prefix to the object name. This helps in tracking objects in buckets, but when you operate on objects you will notice a very different behaviour than operating on files in a traditional file system.
Why bioinformatics needs cloud buckets
There are several features of cloud buckets that make them a near ideal solution for bioinformatics. These include:
- Massive Data Volumes: Bioinformatics often deals with extensive datasets. Genomic sequencing, for instance, can generate terabytes of data. Cloud buckets are designed to scale effortlessly, accommodating everything from a single byte to exabytes, or from billions of objects, to a small handful (traditional file systems struggle to scale efficiently). Moreover, object storage works best with immutable data. Most bioinformatics datasets, like raw reads from a DNA sequencer, are fixed data, and are only ever created, read, and deleted—but never updated.
- Accessibility: Commercially hosted cloud buckets like Google and AWS can be accessed globally without the common bottlenecks of traditional data transfer. Moreover, due to the use of APIs, access is not limited to the constraints of specific OS standards. For example, if you have a traditional unix file system, your group access determines file access, which can be fairly finite. A cloud bucket can allow for multiple access methods to the same file through its API controls, which gives you a lot more granular control.
- Cost-Effective: Running in-house servers and storage systems can be expensive. Cloud buckets often operate on a pay-as-you-go model, ensuring costs are proportional to the size of the data stored. Additionally, most commercial cloud providers provide automated mechanisms to enable cost savings based on use patterns, moving old/stale data to slower but significantly cheaper storage classes, and keeping new or routinely accessed objects in the fastest storage classes. Importantly, there is no upfront hardware cost: this allows even the most resource limited institution to build storage infrastructure without capital expenditures.
- High Durability: Cloud providers typically store multiple copies of data across different locations. This redundancy ensures that even if one datacenter faces issues, the data remains safe and accessible.
- Integrated Analysis Tools: Cloud platforms often offer tools that directly operate on the stored data, from bioinformatics-specific tools to general data analysis frameworks. This integration means data doesn’t need to be moved around – reducing time, cost, and the risk of data corruption. You “bring your analysis” to the data, not the other way around. Additionally, popular workflow management tools for bioinformatics like Cromwell (Terra) and Nextflow (Seqera Tower) support workflows referencing objects stored in different cloud buckets. This is enabled through the use of APIs.
- Automation and Scalability: Bioinformatics can involve repetitive tasks on varying scales. Cloud buckets, combined with other cloud services, allow for automation of these tasks. Whether it’s processing data from ten samples or ten thousand, the system can scale based on demand.
Buckets at work in bioinformatics
Object storage on CLIMB
Object storage serves as the primary data repository for the CLIMB-BIG-DATA platform. CLIMB-BIG-DATA users typically utilise a “notebook server” that provides a lightweight Linux installation that hosts a JupyterLab front-end. The user can upload/download files from the object storage locations in a number of ways.
Internally, the storage system is provisioned using the CEPH storage platform, which offers a robust and scalable solution. CEPH exposes an S3 compatible API, enabling seamless integration with existing tools and software that support the Amazon S3 protocol (nearly all object storage implementations in commercial or private clouds use this protocol). This means that CLIMB-BIG-DATA users can take advantage of a wide range of standard client-side software, such as s3cmd, to interact with their data stored in the platform. The compatibility with the S3 API extends to popular libraries like boto3 in Python. This allows Python developers to effortlessly integrate their data-intensive applications with CLIMB-BIG-DATA, enabling smooth and efficient data management and analysis.
The adoption of the S3 compatible API also benefits users who work with workflow languages like Snakemake and Nextflow. These workflow languages often include built-in support for S3, enabling users to easily integrate the platform into their data processing pipelines. An S3 bucket can also be configured to server files publicly, allowing collaborators (outside of CLIMB) to access static files through a simple URL.
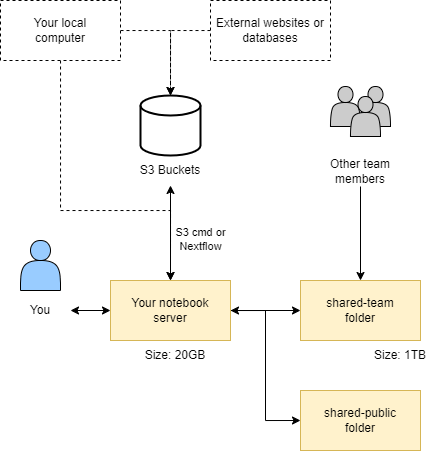
Storing and retrieving files from an S3 bucket is simple with s3cmd, for example, adding a file is:
s3cmd put /path/to/local/file s3://bucket-name/destination/path/While downloading a file would be:
s3cmd get s3://bucket-name/path/to/s3/file /path/to/local/destination/
The Terra.bio Platform
Object storage serves as the primary data repository for the Terra.bio platform as well. Users of the Terra platform enjoy a mature browser-based interface to both their data and workflows. Parameters and variables are entered through the browser window and passed to the Cromwell workflow engine backend for processing on Google Cloud through their Life Sciences API. The Cromwell engine utilises the Google Cloud Storage API. Thus the users need only supply the object address for processing in Terra. This is incredibly useful not only for user data uploaded to Terra workspaces like read data, but also for reference files and data sets.
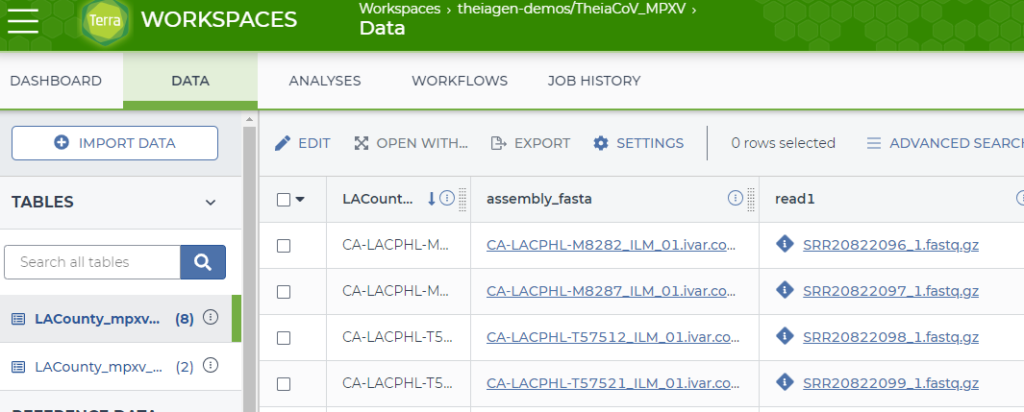
One example can be found in the MPXV workspace that is publicly available on Terra (https://app.terra.bio/#workspaces/theiagen-demos/TheiaCoV_MPXV/data). Figure 2 shows a screenshot of the data table tab. The data table provides a specimen centric view of your analyses with both the input data and results from analysis. Two columns that are shown are “assembly_fasta” and “read1”. They are shown as hyperlinks in the browser but they are actual object addresses that are rendered as hyperlinks. The data for “read1”, in the first row, an input file, is actually:
gs://theiagen-public-files-rp/terra/mpxv-files/featured-workspace-20220819/SRR20822096_1.fastq.gzThis is the GCP object address for the fastq file to be used in the analysis. It is actually hosted outside this workspace in the gs://theiagen-public-files-rp bucket. This allows users to access the single copy of this file without having to replicate the data when they clone the workspace, saving on storage costs.
The assembly_fasta data is a result file that is stored in the workspace bucket. The actual address for the result in the first row is:
gs://fc-4cbb54c1-cea8-4f30-8ebe-81b0a10ab6a1/submissions/d081b57e-1886-415c-aa23-1ce6de6411f9/theiacov_illumina_pe/dcd3b3c9-c03e-41a9-87d0-51ee9283062e/call-consensus/cacheCopy/CA-LACPHL-M8282_ILM_01.ivar.consensus.fastaThe bucket address is gs://fc-4cbb54c1-cea8-4f30-8ebe-81b0a10ab6a1 while the rest of the “virtual path” (a simulated folder structure where the “path” is actually a name prefix, https://cloud.google.com/storage/docs/objects) describes the submission, task and data.
A second screenshot is shown in Figure 3. This is part of the “Workspace Data” associated with the workspace, and is incredibly useful for referencing files that are needed across all specimens in a workflow. In this instance, there are hyperlinks for the MPXV.primer.bed file and the MPXV.MT903345.reference.fasta file. These are found in the gs://theiagen-public-files bucket, with the full object address for the MPXV.primer.bed file gs://theiagen-public-files/terra/mpxv-files/MPXV.primer.bed. This is incredibly powerful because you can replace the reference file and it will impact every workflow that uses that object.
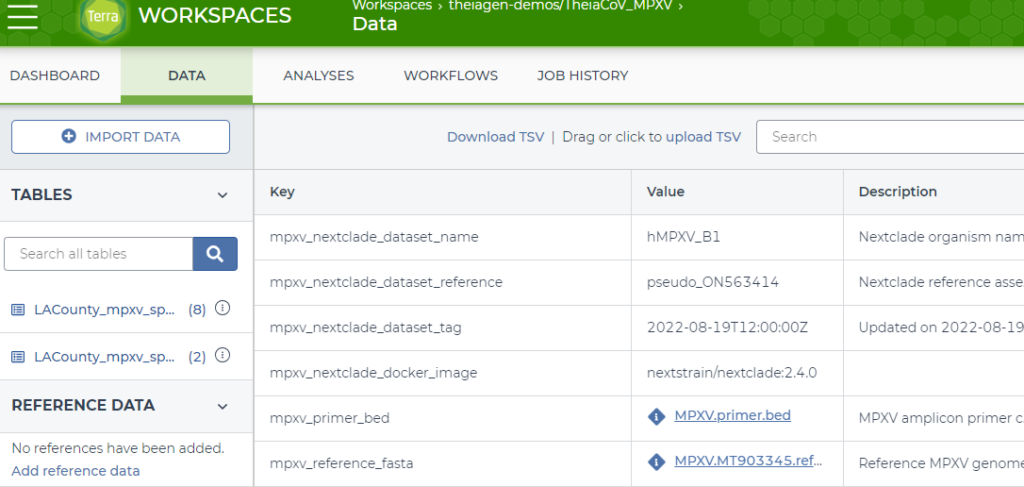
Figure 3: Data tab of the public TheiaCoV_MPXV workspace highlighting workspace data files.
When not to use buckets
You may be thinking “why don’t we use buckets for everything?” Well, there are some downsides to buckets. It was mentioned earlier that buckets work best for immutable data. The converse is true as well: buckets don’t work well for any type of data that needs to be modified or edited. Relational databases are a great example of what NOT to do with buckets. Importantly, most bioinformatic command line scripts still read, write, and modify files in a traditional unix file system as a temporary or ephemeral “working space” for their analyses, while their initial inputs and final outputs are best suited for buckets. Buckets are best suited for “data at rest”.
Also, because of the flat namespace, data in buckets are not inherently organised in any searchable structure, so buckets work best when another system is used to inventory and index the objects in a bucket. The Terra data table in Figure 2 is a good example of this at work.
Are you ready to use buckets?
Although cloud buckets were not created for bioinformatics, it is understandable if you thought they were. Their model of shared access, dynamic scaling, and heterogeneous compute systems, has provided researchers with an environment which allows even the most resource limited institutions to compete with much larger established institutions. One of the most important features of this “bioinformatics equity” is the reliance on cloud buckets by the major data curators like NCBI and EMBL-EBI. Given the migration of most bioinformatics compute resources from custom scripting to the adoption of workflow engines and algorithm containerization, there is a near virtuous interaction between these compute infrastructures and cloud buckets. This became increasingly apparent during the recent pandemic where many tools used for SARS-CoV-2 situation awareness relied on data stored and submitted to cloud buckets.
If you’re not using cloud buckets it may be worth it to learn about them and experiment with them to see how slowly, or quickly you are able to adopt this technology. As bioinformatics progresses, buckets will become more and more essential to most bioinformatics software and infrastructure.


